Updated: Click here for details on Nucro-Technics’ approach to nitrosamine testing.
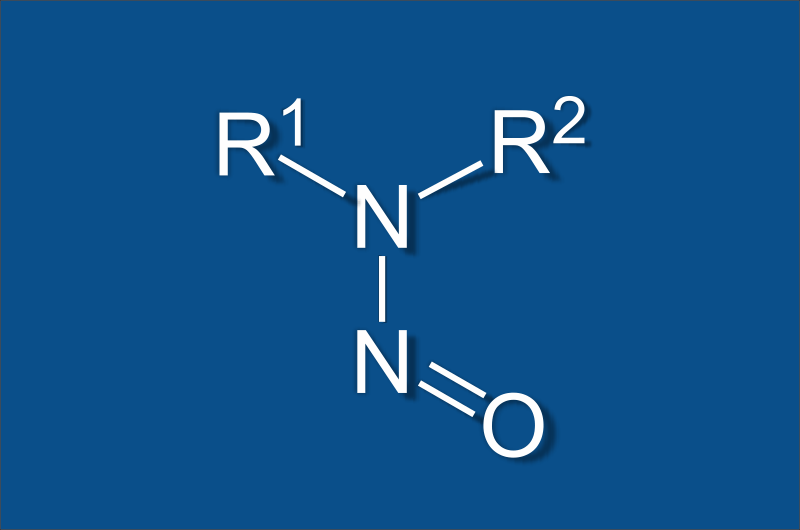
Health Canada recently sent a letter to Market Authorization Holders (MAHs) about nitrosamine testing of impurities in human pharmaceutical products. Nitrosamine are genotoxic compounds that are potentially carcinogenic even at low exposure levels. What the regulators are asking is for the MAHs to perform Nitrosamine testing to evaluate levels of these impurities which include N-nitrosodimethylamine (NDMA), N-nitrosodiethylamine (NDEA), N-nitrosodiisopropylamine (NDIPA), N-nitroso-ethylisopropylamine (NEIPA), and N-nitrosomethyl-n-butylamine (NMBA).
The scope of products that are covered under the Health Canada letter and from further stakeholder discussions are any drug that contains a chemically synthesized active pharmaceutical ingredient – effectively everything except biologics, radiopharmaceuticals, disinfectants and veterinary products. As of today, the products under the most scrutiny right now are Ranitidine (Zantac), Angiotensin Receptor Blockers (Losartan, Candesartan, Irbesartan, Olmesartan, Valsartan, Hyzaar), and Metformin.
Why is Nitrosamine Testing an Issue Now?
The potential for formation of nitrosamine impurities at such low levels during manufacturing processes was not recognized by regulators and the industry until recently. With that being said, nitrosamine impurities may have been present in products for quite some time. In order to actually detect low levels of these impurities, nitrosamine testing requires highly specialized equipment (LC-MS/MS) as well as significant method development expertise. This type of equipment is not typically a part of most GMP laboratories that are involved in release testing of products.
Some additional challenges to this are that detection of nitrosamines is challenging for both regulators and industry. There are multiple potential root causes as well as an expanding scope of impacted products. Nitrosamine impurities which may form during drug product manufacturing also have fewer opportunities to be purged than those which may come about from the drug substance (API) manufacturing process.
What You Need to Do
Market Authorization Holders are required to conduct a thorough and robust risk assessment of their drug products that contain chemically synthesized active pharmaceutical ingredients. An evaluation of whether secondary amines or nitrites coexist during the manufacturing process should be done as well as an evaluation of the potential of contamination through bulk materials and potable water.
Risk assessments should be prioritized based on:
- The maximum daily dose of the product;
- Route of administration;
- Duration of use;
- Use by special populations (such as pregnant women and children);
- Accessible information that suggest an API/drug product class contains nitrosamines; and
- Structural elements in the API or manufacturing details that suggest a higher degree of risk (such as amine groups or the use of nitrites)
Once the risk assessment has been completed, confirmatory nitrosamine testing should be carried out using appropriately validated and sensitive methods.
Any required changes, such as changes to the manufacturing process, or changes to the API or drug product specifications, would then need to be filed.
Health Canada’s current timelines require risk assessments to be completed by October 1, 2020. Confirmatory Testing and any changes to the market authorization must be finalized by October 1, 2022.
How Nucro-Technics Can Help You
Nucro-Technics routinely performs nitrosamine testing using mass spectrometry. We have multiple systems available for the testing of the samples, as well as qualified scientists experienced in the method development and method validation of these tests. Our organization has performed thousands of nitrosamine tests on hundreds of product batches. Because everybody’s needs are different, it is easiest to reach out to us for further information or click here to learn more about Nucro-Technics’ complete range of Mass Spectrometry testing services.